




Hi everybody 👋!
It's been a while since my last post. I've been busy with my PhD🧬, and this post it's related to it. I needed to extract reads (a particular DNA sequence after sequencing). It's a fairly easy process if you have to do it just a couple of times, but I had to analyze 63 Loci (note📙: a locus, in a really simplified explanation, it's a piece of a chromosome, and loci is the plural form of locus), so I needed a way to automatize this task.
Without getting into too much detail, what I did is this: 1) I chose a random loci 2) I tested a way to extract the reads 3) I realized I also needed the number of reeds (the number of times each read appears in the sequencing file) 4) I updated the code from step 2 (with a bit of help from stackoverflow 😎) 5) I applied a for loop in order to repeat the process for all the 63 loci.
Since I run all the bioinformatic scripts I need in Linux 🐧, I wrote this one in shell. Basically, this is the code:
samtools view $origen $locus|cut -f 2,3,4,6,10,14|awk '$4 == "21M"'|sort -k 1|awk -v hebra2=$hebra '$1 == hebra2'|awk 'BEGIN{FS=OFS="\t"} b!=$3{for(j=0;j<i;j++){print a[j],i};delete a;b=$3;i=0;}{a[i++]=$0}END{for (j=0;j<i;j++){print a[j],i}}'|sort -k 6,6|sort -k 3 -n -u|awk -v reads2=$reads '$7 >= reads2 { print }'
Samtools lets you manipulate sequencing data. You have to provide an input file (denoted by the variable $origen here, because it's in Spanish) and a chromosomal location (or simply $locus). The next step it's just for keeping the columns I needed because these files usually have a lot of different columns with information. Using `awk '$4 == "21M"' I force the script to keep only the reads with 21 nucleotides: $4 is the column that gives that information and 21M is the "Flag" for 21 matched nucleotides. After that, I sorted the results and the following chunk of code it's a bit tricky. Essentially, it counts the occurrence of each read and reports it in a new column. The final part just sorts the output and I add a filter in case I would only like to keep sequences with a minimum number of reads.
"Ok, it works! 💪 Time to add a for loop"
for locus in $(cat $file)
do
CODE HERE!!
done
And that's it! Here's an output example:
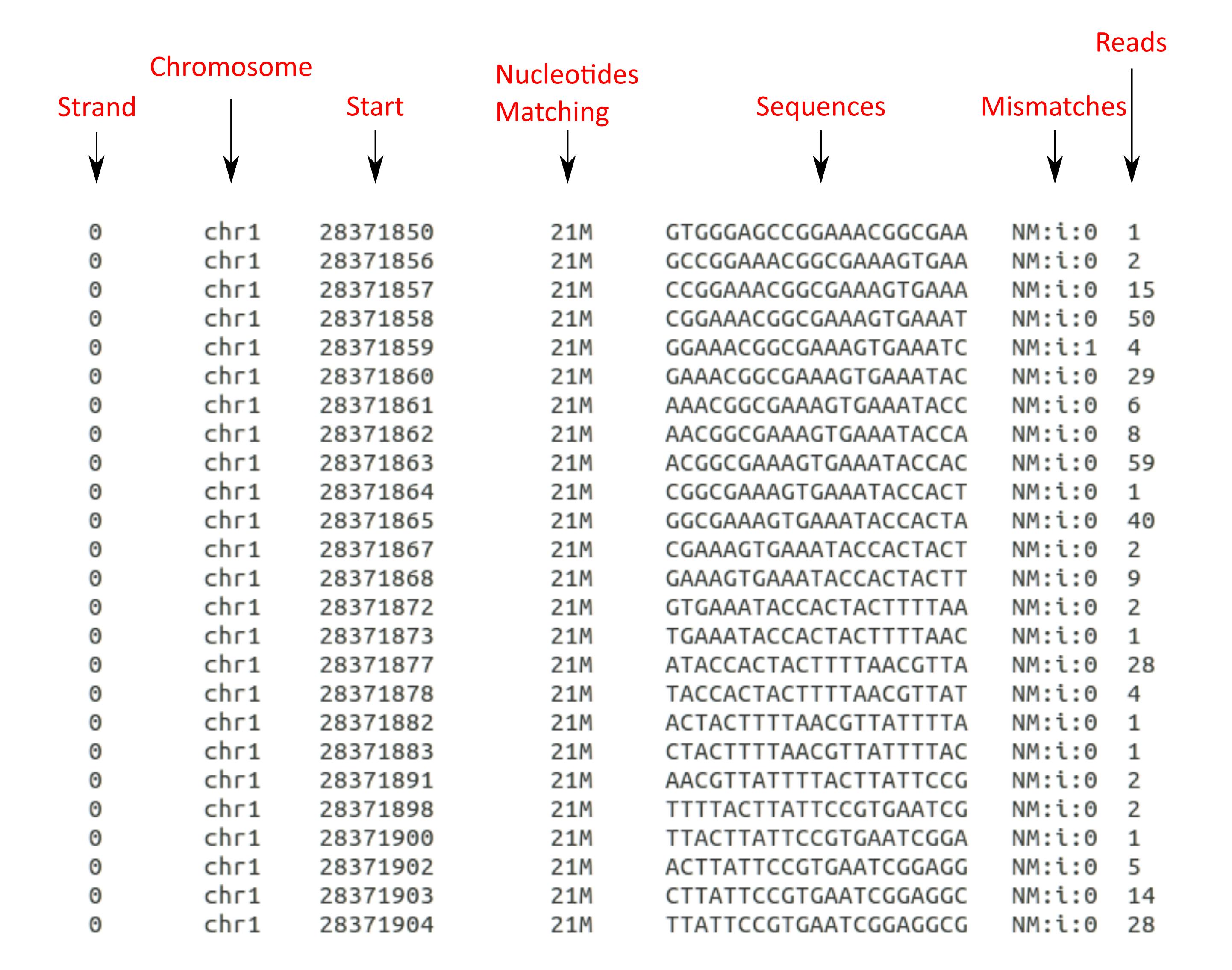
There's something I didn't mention, and it's that sequencing reads can align to both DNA strands (remember DNA it's a double helix? 🧬). I had that in mind when I wrote the script, so I added an option to select the desired strand.
I'm sure this script can be improved, but I'm very happy with the results because it allowed me to speed up my analyzes. I added a few manipulation options, so feel free to check it out Here, it also has a readme file, in case you wanted to use it.
I hope you find this article useful.
Until next time! 🙋♂️
*Cover image was downloaded for free from Unsplash